Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.18 no.3 mar. 2001
revisión de conjunto
Leptina: implicaciones fisiológicas y clínicas
J.I. Botella Carretero, M.D. Lledín Barbancho*, M. a. Valero González, C. Varela Dacosta
Servicio de Endocrinología y Nutrición. Hospital Ramón y Cajal.
*Servicio de Pediatría. Hospital Infantil La Paz. Madrid
RESUMEN
La leptina es una proteína que ha sido identificada hace tres años, pero de cuya acción, o por lo menos déficit, ya se conocía desde el año 1950. Dickie y sus colaboradores comunicaron la aparición en una de sus colonias de ratas, un mutante asociado con obesidad mórbida. El defecto genético se heredaba de forma recesiva y su manifestación era en edad temprana de la vida y se asociaba con diabetes mellitus. En diciembre de 1994 se consiguió clonar el gen ob, lo que constituyó el primer paso para que posteriormente se determinara el producto de dicho gen, la leptina, como una proteína de 167 aminoácidos que se expresaba únicamente en el tejido adiposo. Desde entonces, se ha demostrado la relación de la leptina con numerosos sistemas de regulación neuroendocrina. Los recientes descubrimientos sobre dicha proteína merecían ser puestos al día, así como su posible relevancia presente y futura en la práctica clínica.
PALABRAS CLAVE: Leptina. Obesidad. Diabetes mellitus. Hormonas tiroideas. Suprarrenal. Reproducción. Crecimiento.Leptin: physiologic and clinical roles
ABSTRACT
Leptin is a protein that has been identified three years ago, but its role, or at least its deficiency, was suspected from 1950. Dickie and co-workers reported the appearance of a mutant rat in one of their colonies with morbid obesity. The genetic defect was autosomal recessive and was manifested early in life. In December 1994, the gen ob was cloned, which stated the first step for the later identification of the gen product leptin, as a protein of 167 aminoacids expressed in adipose tissue. Since then, leptin has been implicated in many neuroendocrine regulatory pathways. The recent research in leptin roles worths an update review, and so its current and future clinical relevance.
KEY WORDS: Leptin. Obesity. Diabetes mellitus. Thyroid hormones. Adrenal. Reproduction. Growth.
Trabajo aceptado: 17 de Junio de 2000
Correspondencia: Ignacio Botella Carretero. Hospital Ramón y Cajal. Ctra. Colmenar km 9,700. 28034 Madrid.
INTRODUCCIÓN
Leptina, del griego leptos, que significa delgado, es una proteína que ha sido identificada hace tres años, pero de cuya acción, o por lo menos déficit, ya se conocía desde el año 1950. Sin embargo, ha sido en estos tres últimos años en los que el conocimiento de la misma, de su fisiología y de sus implicaciones clínicas se ha disparado, y su interés científico merece una revisión en el momento actual.
En el año 1950, en el laboratorio Jackson de Bar Harbor, Dickie y sus colaboradores comunicaron la aparición en una de sus colonias de ratas, un mutante asociado con obesidad mórbida. El defecto genético se heredaba de forma recesiva y su manifestación era en edad temprana de la vida y se asociaba con diabetes mellitus. Se denominó ob/ob a ese mutante (1). Poco después, otro tipo de alteración genética en ratones fue descrita por Hummel y Coleman (2), que consistía en la aparición de obesidad y diabetes y que fue denominado db/db. Tras ellos, otras formas fueron descritas y denominadas fatty, tub y fat (3,4). El hecho de que los mutantes ob/ob y db/db fueran fenotípicamente idénticos pero con alteraciones genéticas distintas hizo pensar en la existencia de defectos en distintos pasos de una cadena enzimática aún por definir. De la misma forma, el hecho de que todos ellos tuvieran diabetes mellitus y resistencia insulínica, llevó a estudios extensos sobre actividad insulínica y su receptor. Pero además, faltaba por explicar otras alteraciones observadas en estas ratas tales como hiperfagia, hiperlipidemia, infertilidad e hipotermia.
Mientras tanto, se realizaron estudios que demostraron que la lesión en la zona ventromedial hipotalámica producía obesidad (5). Entre los núcleos implicados en dichos estudios estaban el paraventricular, el central de la amígdala y el ventromedial. Asimismo se demostró que la hiperfagia no era esencial para el desarrollo de obesidad, ni en el ratón ob/ob ni en el modelo hipotalámico, aunque sí era de mayor magnitud cuando la ingesta era libre. Más aún, se implicaron, por una parte una disminución de la actividad simpática sobre la grasa parda y, por otra, que tras la adrenalectomía se producía un retorno a la ingesta normal, masa muscular normal, fin de la resistencia insulínica y de la hiperglucemia, aunque no se corregía la infertilidad. Así surgieron las hipótesis autonómica y endocrina respectivamente (6).
En diciembre de 1994 se consiguió clonar el gen ob, lo que constituyó el primer paso para que posteriormente se determinara el producto de dicho gen (la leptina) como una proteína de 167 aminoácidos que se expresaba únicamente en el tejido adiposo (7,8). Posteriormente se producían comunicaciones sobre la corrección de la obesidad, la diabetes, la infertilidad y la hipotermia en los ratones ob/ob. Sin embargo, los ratones db/db eran completamente resistentes a la acción de la leptina (9-11).
ACCIONES BIOLÓGICAS DE LA LEPTINA
La leptina se produce exclusivamente en las células del tejido adiposo en una gran variedad de especies, incluidos los humanos, y su concentración es mayor en individuos con sobrepeso que en los delgados (12). Recientemente se ha descrito la síntesis de leptina en placenta y en estómago. Sus niveles son mayores en mujeres que en hombres tras ajustar el IMC (13) y existe un ritmo diurno en ambos sexos. El acmé de la leptina ocurre temprano por la mañana y el nadir por la tarde (14). El ritmo de leptina es similar al de la prolactina, TSH, ácidos grasos libres y melatonina (15), e inversamente relacionado con los pulsos de ACTH y cortisol (16). De la misma forma se ha demostrado que los pulsos de leptina son síncronos con los de LH y estradiol en mujeres normales y que la obesidad puede evitar estos pulsos (17). En cuanto a su farmacocinética existen dos pools de leptina: un pool rápido, con una vida media de 3,4 minutos en plasma y un pool más lento, con vida media de 71 min. La leptina se une a múltiples proteínas plasmáticas, incluyendo una forma soluble del receptor de la leptina (Re) y la α-2-macroglobulina (18), y su distribución tisular muestra a los 60 y 180 minutos que el intestino es el que contiene la mayor concentración de leptina, mientras que el hígado, riñón, estómago y pulmón tienen cuatro veces menos. Poco abundante es la concentración en la piel, músculo, corazón y cerebro (19).
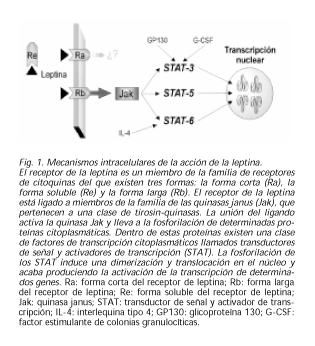
El receptor de la leptina es un miembro de la familia de receptores de citoquinas, que fue clonado y secuenciado por distintos grupos, del que existen tres formas: la forma corta (Ra), la forma soluble del anterior (Re) y la forma larga (Rb) (20). Una mutación glutamina por treonina en el dominio intracelular se identificó en los ratones db/db (21). Otras mutaciones del mismo se encontraron en los otros modelos genéticos de obesidad descritos. Así tenemos que en el ratón fa/fa existe una mutación glutamina por prolina en el dominio extracelular. El ratón fa/fa responde a leptina exógena cuando se inyecta de forma intraventricular por lo que se supone que la alteración está en el transporte de la leptina o en el número de receptores localizados en la membrana plasmática (22). La forma larga del receptor de la leptina (Rb) se ha identificado en múltiples regiones cerebrales y también en tejidos periféricos como el hígado, el páncreas y el músculo estriado. En el cerebro, las regiones fundamentales en las que se ha encontrado se encuentran asociadas a la regulación del comportamiento alimentario y de la regulación y equilibrio energético. En concreto el núcleo arcuato y el ventromedial del hipotálamo. Sin embargo, en el tejido periférico, parece que el receptor predominante es del tipo corto (Ra). En órganos como intestino, pulmón y riñón es donde se encuentran estos receptores los cuales se han implicado en el aclaramiento de la leptina o bien en acciones distintas a las de la activación del Jak-Stat (23). El receptor de la leptina carece de actividad enzimática en su dominio intracelular. En vez de ello, está ligado a miembros de la familia de las quinasas janus (Jak), que pertenecen a una clase de tirosin-quinasas. La unión del ligando activa la quinasa Jak y lleva a la fosforilación de determinadas proteínas citoplasmáticas. Dentro de estas proteínas existen una clase de factores de transcripción citoplasmáticos llamados transductores de señal y activadores de transcripción (STAT). Se han identificado seis miembros de esta familia. La fosforilación de los STAT induce una dimerización y translocación en el núcleo y acaba produciendo la activación de la transcripción de determinados genes. En concreto, la acción de la leptina es capaz de activar los STAT 3, 5 y 6. Previamente descrito, el STAT-3 también se activa por gp130, G-CSF-R y otros. El STAT-6 es también activado por la acción de la IL-4. La activación del STAT-5 no se produce por ninguna otra sustancia conocida (23,24) (Fig. 1).
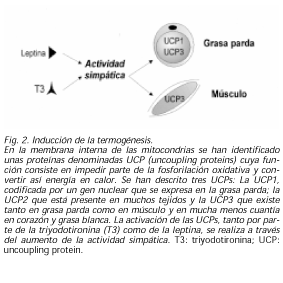
Un importante componente del gasto energético es la termogénesis. En los mamíferos pequeños y en los jóvenes mamíferos grandes, esta actividad se realiza por la grasa parda. En la membrana interna de las mitocondrias de la misma se han identificado unas proteínas de membrana, denominadas UCP (uncoupling proteins) cuya función consiste en transportar protones para equilibrar los mismos a ambos lados, impidiendo parte de la fosforilación oxidativa y convertir así energía en calor. Hasta la fecha se han descrito tres UCPs: La UCP1, codificada por un gen nuclear se expresa sólo en la grasa parda; la UCP2 que está presente en muchos tejidos y la UCP3 que existe tanto en grasa parda como en músculo y en mucha menos cuantía en corazón y grasa blanca. Tanto las hormonas tiroideas como los estímulos β-adrenérgicos son capaces de aumentar la actividad de estas proteínas. Más aún, se ha demostrado que la leptina es capaz también de producir esta activación. En todo caso, parece que la activación de las UCPs, tanto por parte de la T3 como de la leptina, se realiza a través del aumento de la actividad simpática (26,27) (Fig. 2).
En 1998 se ha descrito que los niveles de zinc son necesarios para mantener los niveles séricos de leptina. El zinc parece que produce un aumento de la producción de IL-2 y TNF-α , lo cual parece que aumenta los niveles de leptina (28). De hecho, el TNF-α se expresa de forma constitutiva por los adipocitos y tanto los ratones ob/ob como las ratas fa/fa tienen aumento del TNF-α . Los niveles del mismo se han demostrado elevados en obesos con respecto a individuos normales, y se ha implicado en la resistencia insulínica (29). También la IL-1 se ha implicado en la inducción de leptina, bien directamente o indirectamente a través del eje hipotálamo-hipófiso-adrenal (30). Asimismo se ha demostrado recientemente que tanto la placenta como el epitelio gástrico son tejidos en donde se produce leptina. El significado en el primero podría ser el del desarrollo intrauterino y neonatal (31) y en el segundo podría jugar un papel en los efectos precoces mediados por la CCK, que son inducidos por la ingesta, tales como la saciedad (32).LEPTINA Y OBESIDAD
Inicialmente, tras el descubrimiento de la leptina como producto del gen ob, existió una explicación rápida y sencilla de la función de la misma: se trataba de una hormona anti-obesidad y su falta de producción o su resistencia llevaban al sobrepeso. Sin embargo, con el conocimiento posterior de más detalles sobre la fisiología de la misma, parecía más lógica la llamada teoría del genotipo económico. Esta consistiría en que a lo largo de la evolución, la selección natural habría favorecido un sistema que protegiera al organismo de grandes períodos de ayuno, cosa que ha sido lo más habitual a lo largo de la historia hasta hace poco, e incluso es así en la actualidad para la mayoría de la humanidad (33). El concepto de genotipo económico se introdujo por primera vez en 1963 con la propuesta de que una secreción rápida de insulina favorecería el rápido almacenamiento de nutrientes por los tejidos pero que, en un aporte libre de grandes cantidades de calorías, ocurriría una resistencia insulínica y una diabetes (34). Es posible que, con los conocimientos actuales sobre la leptina, ésta sea esa señal de suficiencia o insuficiencia de reservas energéticas y que más que una hormona anti-obesidad, haya jugado un papel importante en la evolución del hombre como hormona indicadora de las reservas energéticas.
En el núcleo arcuato hipotalámico se produce NPY. Este neurotransmisor llega a través de las proyecciones de las neuronas de dicho núcleo hasta el núcleo paraventricular, que es la zona donde se libera (35). Existen varios estudios que han demostrado que el aumento de NPY lleva a un aumento de la sensación de hambre y con ello a la hiperfagia y obesidad (36). Existen evidencias de que la leptina actúa a nivel del núcleo arcuato impidiendo la formación de NPY y por tanto, indirectamente evitando la obesidad, y parece que es a través de esta acción de la leptina por la que se produce la activación de las UCPs (37). Sin embargo se ha demostrado en ratones transgénicos que el NPY no es esencial para la acción de la leptina, insinuando que otras vías deben formar parte de su acción (38).
El tejido adiposo, aunque difusamente distribuido, es capaz de secretar diversas sustancias y es fundamental en la regulación y almacenamiento de la energía. Como hemos mencionado, la leptina se produce fundamentalmente en la grasa blanca y existe una relación entre niveles de leptina y depósitos de grasa, que se relacionan con el IMC. Esta relación es curva en vez de completamente lineal, lo que sugiere que los niveles de leptina aumentan de forma exponencial con el aumento de la masa grasa (39). Otros estudios han demostrado que los niveles de leptina son mayores en la grasa subcutánea que en la grasa visceral, siendo los de PPARγ al revés, mientras que el inhibidor celular de la proteína-2 de apoptosis (cIAP2) se expresa más en esta última (40). Parece ser que la grasa visceral expresa más receptores adrenérgicos y es por tanto más sensible a la acción lipolítica adrenérgica y menos sensible a la acción lipogénica de la insulina (41). También se ha demostrado, en concordancia con esto, que la regulación de la grasa subcutánea por parte de la leptina depende de la zona ventromedial talámica, cosa que no ocurre con el contenido graso de los islotes pancreáticos (42). De hecho, la activación de los receptores β3 aumenta la termogénesis pero no afecta a los niveles de NPY ni la expresión de CRH, sugiriendo una vez más que la acción de la leptina se produce a través de otras vías además de la del NPY y que existen diferencias en cuanto a la acción sobre la grasa subcutánea y la visceral y parda (43). Recientemente se ha descrito que la activación de la LPL por medio de la leptina es independiente de la actividad adrenérgica, cosa que no ocurre con la activación de las UCPs (44). Sin embargo la administración aguda de leptina produce hipertrigliceridemia porque interfiere con la captación de triglicéridos por el músculo y el adipocito (45).
Los niveles de leptina no cambian con una sobrecarga de glucosa ni con comidas mixtas. Es tan sólo a partir de 6 horas y más claramente tras 12 horas de ayuno o sobrealimentación cuando se ven cambios. Parece ser, por tanto, que los niveles de leptina se relacionan más con los cambios a largo plazo (46-48). Tanto el exceso de ingesta a largo plazo, como el ayuno prolongado, son capaces de variar los niveles de leptina más allá de lo esperado desde el punto de vista del IMC. Recordemos que, como hemos dicho, comienzan a implicarse micronutrientes como el zinc en la regulación de los niveles de leptina (ver arriba). Además se está comenzando a implicar a la leptina como responsable de ciertas preferencias alimenticias. Este es el caso de un estudio en el que parece que los niveles elevados de leptina en mujeres obesas se asocian con disminución de la ingesta total de calorías y una disminución de la preferencia por las grasas. Esto sin embargo requiere confirmación en otros estudios (49).
El descubrimiento de la leptina y el hecho de que su administración a ratones ob/ob evitara la obesidad, sentó la esperanza de la posible solución de la obesidad humana si es que ésta era por un déficit de leptina. Sin embrago, es excepcional este hecho en la obesidad humana, habiéndose comunicado muy pocos casos de déficit congénito de leptina (50). Por el contrario, la obesidad humana parece un estado de resistencia a leptina en la mayoría de los casos y se está investigando sobre posibles alteraciones a nivel del receptor de la leptina o en otro punto a nivel postreceptor (51,52). En todo caso, se ha descubierto que la capacidad de transporte de la leptina a través de la barrera hematoencefálica (BHE) está disminuida a partir de un cierto nivel umbral de concentración plasmática (53). Por ello, incluso en el caso en que la obesidad resistente a leptina se pudiera tratar con dosis altas de la misma, se debería solucionar este problema del transporte a través de la BHE (54). Más aún, se ha especulado recientemente sobre la posibilidad de que la leptina sirviera como marcador predictivo en cuanto a los cambios de peso. Mientras que parecía que los niveles bajos de leptina en ratones podrían predecir una ganancia de peso, los niveles basales de leptina en humanos con obesidad moderada no tienen valor predictivo alguno sobre el cambio de peso en el futuro (55).
LEPTINA Y HORMONAS TIROIDEAS
Existe una relación lógica entre leptina y hormonas tiroideas: sabemos que la administración de leptina aumenta la actividad simpática sistémica y en el tejido adiposo y el músculo produciendo un aumento de la termogénesis (56). De la misma forma, las hormonas tiroideas son un factor principal en la regulación del metabolismo basal, de la termogénesis y de la actividad simpática. Como ya se ha comentado más arriba, tanto las hormonas tiroideas como la leptina aumentan la actividad de las UCPs y con ello favorecen la termogénesis. Igualmente parece que las hormonas tiroideas pueden tener un papel en la regulación y producción de leptina por los adipocitos (57), posiblemente inhibiendo sus niveles (58).
La leptina puede inhibir directamente la producción de glucocorticoides en las suprarrenales y, dado que los corticoides producen un efecto directo sobre las células del núcleo paraventricular reduciendo los niveles de proTRH, el aumento de los niveles de leptina puede aumentar, de forma indirecta, la actividad tiroidea (59). Por otro lado, la leptina produce, como se ha dicho antes, una inhibición de la producción de NPY en el núcleo arcuato lo cual aumentaría también la producción de TRH (60). Recientemente se ha demostrado que durante el ayuno, el núcleo arcuato es esencial para la normal respuesta homeostática del eje HHT y que es un área crítica en la que median las acciones de la leptina. De hecho, al administrar leptina a ratas en ayuno, se normalizan los bajos niveles de T4 y proTRH encontrados durante el mismo, pero si se lesiona el núcleo arcuato no se produce esta normalización y además en estas ratas, no se produce reducción de hormonas tiroideas en el ayuno (61). En resumen, parece que en los estados de ayuno, lo que interesa es el ahorro de energía y por ello, el aumento de cortisol y la disminución de la leptina producirían una disminución de los niveles de TRH, tanto por acción directa del cortisol a nivel central como por el aumento del NPY hipotalámico. Por el contrario, en un estado de abundancia calórica, al aumentar los niveles de leptina y disminuir los de cortisol, se produciría un aumento de la termogénesis y del metabolismo basal, tanto por acción de la leptina y hormonas tiroideas sobre las UCPs como por la disminución del NPY hipotalámico (Fig. 3).
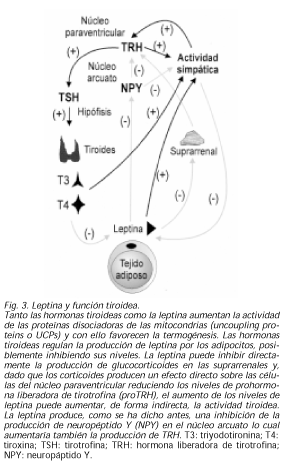
LEPTINA Y EJE HIPOTÁLAMO-HIPÓFISO-ADRENAL
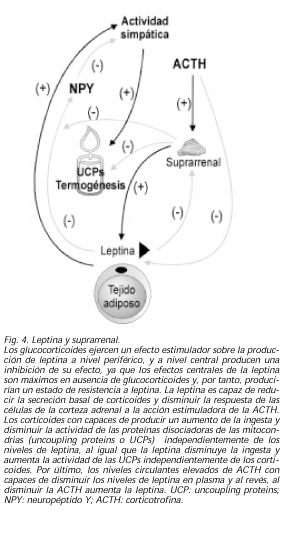
Como hemos mencionado antes, la primera relación observada entre leptina y las suprarrenales fue el hecho de que en ratones ob/ob, tras la adrenalectomía se producía un retorno a la ingesta normal, masa muscular normal, fin de la resistencia insulínica y de la hiperglucemia, aunque no se corregía la infertilidad. Desde esta observación inicial se ha investigado sobre la asociación entre leptina y eje hipotálamo-hipófiso-adrenal (HHA) y sus posibles implicaciones fisiológicas y clínicas. La primera observación fue que en estudios tanto in vitro como in vivo, en ratones y en humanos, quedó demostrado que los glucocorticoides aumentaban la transcripción de leptina y sus niveles plasmáticos (62,63). Recientemente esto se ha confirmado, llegándose a demostrar incluso que en los pacientes con síndrome de Cushing los niveles de leptina están aumentados (64). Pero además, no sólo los glucocorticoides ejercen un efecto estimulador sobre la producción de leptina a nivel periférico, sino que parece que a nivel central producen una inhibición de su efecto, ya que los efectos centrales de la leptina son máximos en ausencia de glucocorticoides y, por tanto, producirían un estado de resistencia a leptina (65). Y a la inversa, la leptina es capaz de reducir la secreción basal de corticoides y disminuir la respuesta de las células de la corteza adrenal a la acción estimuladora de la ACTH (66). En este último año, se ha demostrado que los corticoides con capaces de producir un aumento de la ingesta y disminuir la actividad de las UCPs independientemente de los niveles de leptina, al igual que la leptina disminuye la ingesta y aumenta la actividad de las UCPs independientemente de los corticoides. Esto no quiere decir que no exista relación entre estas hormonas, sino que demuestra que además de existir una interdependencia entre las mismas, ambas también poseen efectos directos muy importantes sobre el control energético (67).
Ya desde los descubrimientos anteriores se pensó en la posible existencia de un mecanismo de relación entre la leptina y el eje HHA incluso a nivel central. Recientemente se ha publicado que los niveles circulantes elevados de ACTH son capaces de disminuir los niveles de leptina en plasma y al revés, al disminuir la ACTH aumenta la leptina (68) (Fig. 4). Se ha demostrado en ratas que la alimentación rica en grasas produce una facilitación de la respuesta al estrés dependiente del eje HHA. Pues bien, durante el período neonatal, la ingesta rica en grasa de la leche materna podría aumentar los niveles de leptina y a su vez disminuir la activación del eje HHA. Tras suspender la lactancia, disminuirían los niveles de leptina, permitiendo la facilitación de las respuestas de estrés ACTH dependientes, como se observan en los adultos con dietas grasas (69), lo que permitiría una cierta protección de los neonatos a la excesiva respuesta al estrés.
LEPTINA Y HORMONA DE CRECIMIENTO
La secreción pulsátil de GH es muy sensible a las alteraciones en el estado nutricional, pero los mecanismos de este hecho son desconocidos. Así en la desnutrición se pierde la intensidad de los picos de GH, alterando con ello (y otros factores) el crecimiento. Los receptores de la leptina (Rb) son abundantes en los núcleos hipotalámicos involucrados en la regulación de GH sugiriendo de la leptina podría ser un factor regulador importante. Pues bien, en estudios recientes se ha visto que la leptina es capaz de favorecer la liberación de GH y que el mecanismo sería en parte debido a la inhibición hipotalámica de la somatostatina (70).
Las implicaciones clínicas no son menos impresionantes: en el año 1998, Kristom y col. han demostrado que los cambios en los niveles de leptina sérica a los tres meses de iniciar el tratamiento con GH era la variable más importante para explicar la variabilidad en la respuesta a este tratamiento (71). Ello abre la puerta a dar una explicación científica, a la vez que un parámetro de pronóstico excelente, a la enorme variabilidad de la respuesta al tratamiento con GH.
LEPTINA Y REPRODUCCIÓN
El primer dato que relaciona la leptina con la función reproductora es el hecho de que los ratones ob/ob, recuperan la fertilidad tras el tratamiento con leptina. Desde entonces se han demostrado muchas relaciones entre leptina y hormonas sexuales: desde las diferencias entre varones y mujeres mencionadas antes en cuanto a los niveles de leptina hasta su implicación en el desarrollo puberal. Se sugirió en un principio que las diferencias sexuales en cuanto a los niveles de leptina se podrían deber a los efectos inductores de los gestágenos o estrógenos y/o al posible efecto supresor de los andrógenos en cuanto a la producción de leptina (72) o por resistencia periférica (73). De hecho, los estrógenos son capaces de aumentar in vivo la producción de leptina en humanos (74) y además producen una alteración de la relación entre la forma larga y corta del receptor, aumentando la sensibilidad tisular por la leptina (75). Incluso se ha establecido una relación entre las conductas alimentarias que se observan con los cambios estrogénicos (la ovariectomía produce un aumento de la ingesta y del peso) y la leptina (76,77). Sin embargo, en estudios recientes, parece que la relación entre estrógenos y leptina se debe a acciones indirectas por otras vías más que por acción directa. Así en un estudio se ha demostrado que los niveles de leptina en las mujeres estaban relacionados con los niveles de catecolaminas durante los ciclos pero no con los niveles de estrógenos ni progestágenos (78) y en otro estudio que la acción de la leptina sobre el sistema reproductor es a través de sus acciones en el metabolismo oxidativo (79). Claramente se necesitan más estudios.
Existen hoy en día datos claros de la relación existente entre leptina y LH. La administración de leptina durante la desnutrición mejora la función reproductora. Esto se debe a que la leptina es capaz de prevenir la supresión de los pulsos de LH, incluso en ausencia de estrógenos, lo que implica una actividad directa y central de la leptina sobre las gonadotrofinas o sobre GnRH (80,81). Como ya hemos dicho, la placenta es un órgano productor de leptina. Durante la gestación no se han encontrado cambios en la concentración de leptina más allá de la esperada por el IMC entre la 18 y la 35 semanas. Sin embargo, ya desde el momento del nacimiento, las diferencias sexuales entre varón y mujer observadas en el adulto en cuanto a la concentración de leptina, se encuentran en las tomas de sangre de cordón (82). Los niveles de leptina son paralelos al peso del recién nacido y también disminuyen en los hijos de madres fumadoras (83).
Se ha propuesto que la leptina podría servir como señal del nivel de masa grasa crítica para la iniciación de la pubertad y mantenimiento de los ciclos reproductores. En apoyo de esta hipótesis está el descubrimiento reciente de un tipo de hipogonadismo hipogonadotropo por mutaciones del receptor de la leptina (84). También se han descrito niveles elevados de leptina en pacientes con pubertad precoz central (85), lo que supone un dato más que apoya la hipótesis de la leptina como señal para el comienzo de la edad reproductora. Además de lo comentado en cuanto a pubertad, la leptina parece implicada en enfermedades que afectan a la reproducción tales como el síndrome de ovario poliquístico (SOP). Las mujeres con SOP son hiperandrogénicas y la mayoría se caracterizan por hiperinsulinemia, resistencia a la insulina y obesidad, particularmente con el fenotipo visceral. También se caracterizan por niveles elevados de leptina circulante. Si bien en un estudio se demostró que estos niveles elevados se relacionaban con el IMC (86), en otros se vio que no dependían del IMC ni del hiperandrogenismo ni de la hiperinsulinemia, aunque sí del patrón de distribución grasa (87). Recientemente en otro estudio se ha conseguido disminuir los niveles de leptina tras el tratamiento con diazóxido en mujeres con SOP (88). Al estudiar los niveles de leptina en mujeres hirsutas, se objetivó que sus niveles no se relacionaban con el hirsutismo, sino con la obesidad y resistencia insulínica (89).
LEPTINA Y PÁNCREAS ENDOCRINO
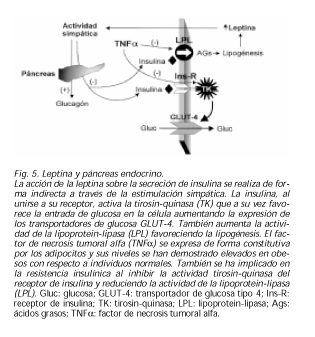
Ya desde un principio fue clara la relación entre el gen ob/ob y la insulina, pues los ratones obesos con este gen tenían también diabetes mellitus y lo mismo pasaba con los ratones fa/fa que tenían alterado el receptor de la leptina. La insulina es capaz de aumentar la producción de leptina en ratas y en los adipocitos humanos in vitro (90). Sin embargo, y aunque la insulina tiene influencia sobre los niveles de leptina en humanos a largo plazo (91), la hiperinsulinemia aguda no ha demostrado gran efecto en humanos, aunque sí en ratas (92). Ello podría tener el significado de que la hiperinsulinemia crónica produce lipogénesis y con ello aumento de la masa grasa total con lo que aumentarían los niveles de leptina secundariamente. Por otro lado, también se han investigado los efectos de la leptina sobre la insulina y recientemente se ha demostrado que en ratas con peso normal, la hiperleptinemia aguda no produce resistencia insulínica (93). Sin embargo, sí es cierto que en ratas vagotomizadas, la respuesta insulínica a la sobrecarga con glucosa es menor cuando se les administra leptina, sugiriendo un mecanismo de inhibición de la secreción de insulina. Pero tanto en ratas normales como en ratas vagotomizadas y simpatectomizadas a la vez, no se encontró este efecto. Ello demuestra que la acción de la leptina sobre la secreción de insulina se realiza de forma indirecta a través de la estimulación simpática (94).
Tanto la resistencia insulínica como la diabetes se corregía en ratones ob/ob tras la administración de leptina. Ello llevó a buscar en los individuos diabéticos posibles mutaciones en el gen de la leptina. Sin embargo el estudio de esta región genética de forma masiva no demostró dicha asociación (95). Además en varios estudios se vio que los niveles de leptina son iguales en individuos con diabetes tipo 2 que en controles ajustando a valores de IMC (96). Es más, el tratamiento con troglitazona en estos pacientes no demostró tampoco variación en los niveles de leptina tras ajustar por IMC (97). Igualmente en la diabetes tipo 1, aunque inicialmente parecía que tenían mayores niveles de leptina que los controles (98), se ha demostrado recientemente que las concentraciones de leptina en estos pacientes son similares a las de personas sanas y que la asociación existente real es entre leptina y obesidad (99). Por otro lado, la distribución de grasa se ha relacionado con la resistencia insulínica y se especuló sobre la posibilidad de un papel de la leptina en ello. En un estudio reciente se ha visto que los niveles de leptina se relacionan inversamente con los de ácidos grasos libres y no con la distribución de la grasa corporal (100). La secreción de glucagón tras la hipoglucemia se ve claramente facilitada por la inyección de leptina en ratas. Cuando se realiza este experimento en ratas simpatectomizadas este efecto no se observa. Una vez más, parece que el efecto de la leptina sobre el glucagón, al igual que lo que ocurre con la insulina, depende de la activación del sistema simpático (101).
El TNFα se expresa de forma constitutiva por los adipocitos y tanto los ratones ob/ob como las ratas fa/fa tienen aumento del TNFa. Los niveles del mismo se han demostrado elevados en obesos con respecto a individuos normales, y se ha implicado en la resistencia insulínica (102). En cuanto al mecanismo parece ser que existe una inhibición de la actividad tirosin-quinasa del receptor de insulina disminuyendo la expresión de los transportadores de glucosa GLUT-4. Igualmente el TNFα reduce la actividad de la LPL, estimula la lipolisis hepática y aumenta el contenido de PAI-1 en adipocitos. La dieta disminuye los niveles de TNFa y de leptina sérica y se asocia a una mejora en la sensibilidad insulínica y del metabolismo lipídico. La edad aumenta los niveles de TNFα y se relaciona con aumento de la resistencia insulínica (103,104) (Fig. 5).
LEPTINA Y OTRAS ENTIDADES
Alteraciones del comportamiento alimentario: Se ha visto que los niveles de leptina en la anorexia nerviosa y en la bulimia son similares a los de los controles ajustando por IMC. Sin embargo, es posible que la leptina tenga utilidad en la anorexia nerviosa ya que sus niveles se normalizan antes que la ganancia de peso durante el tratamiento, tanto en plasma como en líquido cefalorraquídeo, aunque se ha especulado con la posibilidad de que el aumento precoz de la misma y la dificultad para ganar peso podría representar un cierto estado de resistencia a la leptina (105,106).
Insuficiencia renal crónica: Estos pacientes tienen un aumento de los niveles de leptina, lo cual puede deberse a la disminución de aclaramiento renal. Sin embargo también es posible que la uremia tenga efectos directos sobre la leptina, aunque claramente se necesitan más estudios para confirmar esto y para ver las implicaciones de la hiperleptinemia en la IRC (107).
Infección por VIH: Se ha demostrado que, a pesar de que los pacientes con caquexia asociada tienen un IMC menor que los controles, los niveles de leptina sérica eran similares, lo que puede implicar un estado de hiperleptinemia posiblemente debido a la activación del sistema inmune y a la producción de diversas citoquinas (108). Su significado o implicaciones clínicas se desconocen por el momento. Recientemente se ha descrito un síndrome de lipodistrofia con hiperlimia asociado al tratamiento de la infección VIH con inhibidores de la proteasa. Estos pacientes, al contrario que en la caquexia, se ha visto que los niveles de leptina están disminuidos con respecto a los controles. Es posible que la leptina pudiera ser un parámetro más para diferenciar la caquexia de la lipodistrofia y así evitar el tratamiento con anabolizantes en estos últimos, ya que podría empeorar la resistencia insulínica que presentan los pacientes con lipodistrofia y desencadenar o empeorar una diabetes mellitus (109).
Hematopoyesis y linfopoyesis: Debido a la estructura similar de la leptina con otras citoquinas y al hecho de que su receptor pertenezca misma familia de receptores, se ha propuesto un papel regulador de la leptina en la hemo y linfopoyesis. De hecho se ha demostrado que las células de los órganos hematopoyéticos fetales expresan receptores para la leptina (110) y en adultos parece estimular la linfopoyesis y la eritropoyesis (111), aunque se desconoce si estos efectos son endocrinos o paracrinos.
CONCLUSIÓN
Las múltiples implicaciones fisiológicas y clínicas de la leptina están empezando a descubrirse. Si bien su utilidad clínica en el momento actual no está determinada y el potencial terapéutico de la leptina es poco claro, los descubrimientos que la investigación sobre la leptina está trayendo consigo son prometedores en cuanto a futuras terapias, como es el caso de las UCPs y la termogénesis y otras más. En todo caso, el tiempo y la investigación dirán si el esfuerzo merece la pena.
BIBLIOGRAFÍA
1. Ingalls AM, Dickie MD, Snell GD. Obese, new mutation in the mouse. J Hered 1950; 41: 317-318. [ Links ]
2. Coleman DL. Obese and Diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia 1978; 14: 141-148. [ Links ]
3. Zucker LM, Zucker TF. Fatty, a new mutation in the rat. J Hered 1961; 52: 275-278. [ Links ]
4. Coleman DL, Eicher EM. Fat and Tub: two autosomal recessive mutations causing obesity syndromes in the mouse. J Hered 1990; 81: 424-427. [ Links ]
5. Hetherington AW, Ranson SW. Hypothalamic lesions and adiposity in the rat. Anat Rec 1940; 78: 149-172. [ Links ]
6. Bray GA, York DA. Hypothalamic and genetic obesity in experimental animals: an autonomic and endocrine hypothesis. Physiol Rev 1979; 59: 719-809. [ Links ]
7. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425-432. [ Links ]
8. Ogawa Y, Masuzaki H, Isse N et al. Molecular cloning or rat obese cDNA and augmented gene expression in genetically obese Zucker fatty rats. J Clin Invest 1995; 96: 1647-52. [ Links ]
9. Pelleymounter MA, Cullen MJ, Baker MB, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 1995; 269: 540-543. [ Links ]
10. Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse ob protein: evidence for a peripheral signal linking adiposity and central neural networks. Science 1995; 269: 546-549. [ Links ]
11. Halaas JL, Gajiwala KS, Maffei M, et al. Weight reducing effects of the plasma protein encoded by the obese gene. Science 1995; 269: 543-546. [ Links ]
12. Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 1996; 334: 292-295. [ Links ]
13. Schrauwen P et al. Effect of diet composition on leptin concentration in lean subjects. Met 1997; 46: 420-424. [ Links ]
14. Sinha MK et al. Nocturnal rise of leptin in lean, obese and non-insulin dependent diabetes mellitus subjects. J Clin Invest 1996; 97: 1344-1347. [ Links ]
15. Matkovic et al. Gain in body fat is inversly related to the nocturnal rise in serum leptin levels in young females. J Clin Endocrinol Metab 1997; 82: 1368-1372. [ Links ]
16. Licinio J et al. Human leptin levels are pulsatile and inversely related to pituitary-adrenal function. Nat Med 1997; 3: 575-579. [ Links ]
17. Licinio J et al. Synchronicity of frecuently sampled 24 hour concentrations of circulating leptin, luteinizing hormone and estradiol in healthy women. Pro Nat Acad Sci 1998; 95: 2541-2546. [ Links ]
18. Birkenmeter et al. Human leptin forms complexes with alpha-2-macroglubulin wich are recognized by the a2m receptor/LDL receptor-related protein. Eur J End 1998; 139: 224-230. [ Links ]
19. Hill RA, Margetic S, Pegg GG, Gazzola C. Leptin: its pharmacokinetics and tissue distribution. Int J Obes Rel Met Dis 1998; 22: 765-70. [ Links ]
20. Tartaglia LA, Dembski M, Weng X, et al. Identification and expression cloning of a leptin receptor, ob-r. Cell 1995; 83: 1263-1271. [ Links ]
21. Chen H, Charlat O, Tartaglia LA et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996; 84: 491-495. [ Links ]
22. Chua SC, White DW, Wu-Peng XS et al. Phenotype os fatty due to Gln269Pro mutation in the leptin receptor. Diabetes 1996; 45: 1141-1143. [ Links ]
23. Cumin F, Baum HP, et al. Leptin is cleared from the circulation primarly by the kidneys. Int J Obes Rel Met Dis 1996; 20: 1120-1126. [ Links ]
24. Ghilardi N, Ziegler S, Wiestner A, Stoffei R, et al. Defective STAT signaling by the leptin receptor in diabetic mice. Proc Natl Acad Sci USA 1996; 93: 6231-6235. [ Links ]
25. Vaisse C, Halaas JL, Horvath CM, Darnell JE, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wildtype and ob/ob mice but not db/db mice. Nat Genet 1996; 45: 1446-1450. [ Links ]
26. Da-Wei, Yufang, Karas, Reitman. Uncoupling protein-3 is a mediator of thermogenesis regulated by thyroid hormone, beta-3-adrenergic agonist and leptin. J Biol Chem 1997; 272: 24129-24132. [ Links ]
27. Gimeno RE, Dembski M, et al. Cloning and characterization of an uncoupling protein homolog. Diabetes 1997; 46: 900-906. [ Links ]
28. Mantzoros CS, Prasad AS, Beck FW, Grabowski S et al. Zinc may regulate serum leptin concentrations in humans. J Am Coll Nutr 1998; 17: 270-275. [ Links ]
29. Dandona P, Weinstock R, et al. Tumor necrosis factor-a in sera of obese patients: fall with weight loss. J Clin Endocrinol Metab 1998; 83: 2907-2909. [ Links ]
30. Saraf P, Frederich R, Turner E et al. Multiple cytokines and acute inflamation raise mouse leptin levels: potential role in inflammatory anorexia. J Exp Med 1997; 85: 171-175. [ Links ]
31. Hassink et al. Placental leptin: an important new growth factor in intrauterine and neonatal development? Ped 1997; 100: E1. [ Links ]
32. Bado A, Levasseur S, et al. The stomach is a source of leptin. Nature 1998; 20: 790-793. [ Links ]
33. Flier JS. What´s in a name? In search of leptin´s physiologic role. J Clin Endocrinol Metab 1998; 83: 1407-1413. [ Links ]
34. Neel J. Diabetes mellitus: a thrifty genotype rendered detrimental by progress. Am J Hum Gen 1963; 14: 353-362. [ Links ]
35. Leibowitz SF. Specificity of hypothalamic peptides in the control of behavioral and physiological processes. Ann NY Acad Sci 1994; 739: 12-35. [ Links ]
36. Stanley BG et al. NPY chronically injected into the hypothalamus: a powerful neurochemical inducer of hyperphagia and obesity. Peptides 1986; 7: 1189-1192. [ Links ]
37. Kotz CM et al. Neural site of leptin influence on NPY signalling pathways altering feeding and uncoupling protein. Am J Physiol 1998; 275: R478-84. [ Links ]
38. Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking NPY. Nature 1996; 382: 307. [ Links ]
39. Considine et al. Serum immunoreactive leptin concentrations in humans. N Engl J Med 1996; 334: 292-295. [ Links ]
40. Montague CT et al. Depot related gene expression in human subcutaneous and omental adipocytes. Diabetes 1998; 47: 1384-91. [ Links ]
41. Montague CT, Prons JB et al. Depot and sex specific differences in human leptin mRNA expression. Diabetes 1997; 46: 342-347. [ Links ]
42. Koyama et al. Resistance to adenovirally induced hyperleptinemia in rats. J Clin Invest 1998; 102: 728-733. [ Links ]
43. Savontaus et al. Effects of ZD7114, a selective beta-3-adrenoceptor agonist on neuroendocrine mechanisms controlling energy balance. Eur J Phar 1998; 347: 265-74. [ Links ]
44. Scarpace PJ, Matheny M. Leptin induction of UCP1 gene expression is dependent on sympathetic innervation. Am J Physiol 1998; 275: E259-64. [ Links ]
45. López Soriano et al. Short term effects of leptin on lipid metabolism in the rat. FEBS Lett 1998; 431: 371-4 [ Links ]
46. Kolaczynski et al. Responses of leptin to short term fasting and refeeding in humans. Diabetes 1996; 45: 1511-1515. [ Links ]
47. Kolaczynski et al. Responses of leptin to short term fasting and refeeding in humans. Diabetes 1996; 45: 1511-1515. [ Links ]
48. Dallongeville et al. Short term response of circulating leptin to feeding and fasting in man: influence of circadian cycle. Int J Obes Relat Met Dis 1998; 22: 728-33. [ Links ]
49. Karhunen et al. Serum leptin, food intake and preferences for sugar and fat in obese women. Int J Obes Rel Met Dis 1998; 22: 819-21. [ Links ]
50. Montague et al. Congenital leptin deficiency is associated with severe early-onset obesity. Nature 1997; 387: 903-908. [ Links ]
51. Sanz Paris A, Guallar Labrador AM, Albero Gamboa R. Leptina en la endocrinología de la obesidad. An Med Interna (Madrid) 1999; 16: 530-540. [ Links ]
52. Wurtman RJ. What is leptin for and does it act on the brain? Nat Med 1996; 2: 492-493. [ Links ]
53. Schwartz MW, et al. Cerebrospinal fluid leptin levels: relationship to plasma leptin levels and to adiposity in humans. Nat Med 1996; 2: 589-593. [ Links ]
54. Caro et al. Decreased cerebrospinal fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet 1996; 348: 159-161. [ Links ]
55. Haffner SM et al. Leptin concentrations do not predict weight gain. Int J Obes Rel Met Dis 1998; 22: 695-699. [ Links ]
56. Haynes WG, Morgan DA et al. Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest 1997; 100: 270-278. [ Links ]
57. Yoshida et al. Regulation of expression of leptin mRNA and secretion of leptin by thyroid hormone in 3T3-L1 adipocytes. Bio Bio Res Com 1997; 232: 822-826. [ Links ]
58. Escobar-Morreale et al. Thyroid hormones influence serum leptin concentrations in the rat. Endocrinology 1997; 138: 4485-4488. [ Links ]
59. Bornstein SR, Uhlmann K et al. Evidence for a novel peripheral action of leptin as a metabolic signal to the adrenal gland. Diabetes 1997; 46: 1509-1511. [ Links ]
60. Orban Z, Bornstein SR, Chrousos GP. The interaction between leptin and the hypothalamic-pituitary-thyroid axis. Hor Met Res 1998; 30: 231-235. [ Links ]
61. Legradi G, Emerson CH, et al. Arcuate nucleus ablation prevents fasting-induced suppression of proTRH mRNA in the hypothalanmic paraventricular nucleus. Neuroendocrinology 1998; 68: 89-97. [ Links ]
62. De Vos P, Saladin R, Auwers J, Staels B. Induction of ob gene expression by corticosteroids is accompanied by body weight loss and reduced food intake. J Biol Chem 1995; 270: 15958-61. [ Links ]
63. Murakami et al. Dexamethasone regulates obese expression in isolated rat adipocytes. Bio Bio Res Com 1995; 214: 1260-67. [ Links ]
64. Masuzaki et al. Glucocorticoid regulation of leptin synthesis and secretion in humans: elevated plasma leptin levels in Cushings syndrome. J Clin Endocrinol Metab 1997; 82: 2542-47. [ Links ]
65. Zakrzewska et al. Glucocorticoids as counterregulatory hormones of leptin. Toward an understanding of leptin resistance. Diabetes 1997; 46: 717-719. [ Links ]
66. Bornstein et al. Evidence for a novel peripheral action of leptin as a metabolic signal to the adrenal gland. Leptin inhibits cortisol release directly. Diabetes 1997; 46: 1235-1238. [ Links ]
67. Arvaniti et al. Leptin and corticosterone have opposite effects on food intake and the expression of UCP1 mRNA in brown adipose tissue o ob/ob mice. Endocrinology 1998; 139: 4000-3. [ Links ]
68. Spinedi E, Gaillard RC. A regulatory loop between the hypothalamo-pituitary-adrenal axis and circulating leptin: a physiological role of ACTH. Endocrinology 1998; 139: 4016-20. [ Links ]
69. Trottier et al. Increased fat intake during lactation modifies hypothalamic-pituitary-adrenal responsiveness in developing rat pups: a possible role for leptin. Endocrinology 1998; 139: 3704-11. [ Links ]
70. Tannenbaum GS, Gurd W, Lapointe M. Leptin is a potent stimulator of spontaneous pulsatile growth hormone secretion and the GH response to GHRH. Endocrinology 1998; 139: 3871-5. [ Links ]
71. Kristrom B et al. Short term changes in serum leptin levels provide a strong metabolic marker for the growth response to growth hormone treatment in children. J Clin Endocrinol Metab 1998; 83: 2735-41. [ Links ]
72. Rosenbaum et al. Effects of gender, body composition, and menopause on plasma concentration of leptin. J Clin Endocrinol Metab 1996; 81: 3424-3427. [ Links ]
73. Kennedy et al. The metabolic significance of leptin in humans: gender-based differences in relationship to adiposity, insulin sensitivity and energy expenditure. J Clin Endocrinol Metab 1997; 82: 1293-1300. [ Links ]
74. Saad et al. Sexual dimorphism in plasma leptin concentrations. J Clin Endocrinol Metab 1997; 82: 579-584. [ Links ]
75. Bennet et al. Differential expression and regulation of leptin receptors isoforms in the rat brain: effects of fasting and estrogen. Neuroendocrinology 1998; 67: 29-36. [ Links ]
76. Yoneda et al. The influence of ovariectomy on ob gene expression in rats. Horm Metabol Res 1998; 30: 263-265. [ Links ]
77. Mannuci et al. Relationship between leptin and estrogens in healthy women. Eur J End 1998; 139: 198-201. [ Links ]
78. Mills PJ, Ziegler MG, Morrison TA. Leptin is related to epinephrine levels but not reproductive hormone levels in cycling african-american and caucasian women. Life Sci 1998; 63: 617-623. [ Links ]
79. Schneider et al. Leptin indirectly affects estrous cycles by increasing metabolic fuel oxidation. Horm Behav 1998; 33: 217-228. [ Links ]
80. Nagatani et al. Evidence for GnRH regulation by leptin: leptin administration prevents reduced pulsatile LH secretion during fasting. Neuroendocrinology. 1998; 67: 370-6. [ Links ]
81. Teirmaa et al. Correlation between circulating leptin and LH during the menstrual cycle in normal-weight women. Eur J End 1998; 139: 190-194. [ Links ]
82. Helland et al. Leptin levels in pregnant women and newborn infants: gender differences and reduction during the neonatal period. Ped 1998; 101: E12. [ Links ]
83. Mantzoros et al. Effect of birth weight and maternal smoking on cord blood leptin concentrations. J Clin Endocrinol Metab 1997; 82: 2856-2861. [ Links ]
84. Clement et al. A mutation in the human leptin receptor gene causes obesity and pituitary disfunction. Nature 1998; 392: 398-401. [ Links ]
85. Palmert MR, Radovick S, Boepple PA. Leptin levels in children with central precocious puberty. J Clin Endocrinol Metab 1998; 83: 2260-2265. [ Links ]
86. Rouru et al. Serum leptin concentrations in women with POS. J Clin Endocrinol Metab 1997; 82: 1697-1700. [ Links ]
87. Vicennati et al. Serum leptin in obeses women with POS is correlated with body weight and fat distribution but not with androgen and insulin levels. Met 1998; 47: 988-992. [ Links ]
88. Krassas et al. Leptin levels in women with POS before and after treatment with diazoxide. Eur J End 1998; 139: 184-189. [ Links ]
89. Escobar-Morreale HF et al. Circulating leptin concentrations in women with hirsutism. Fertility and Sterility 1997; 66: 898-906. [ Links ]
90. Rentsch J, Chiest M. Regulation of the ob mRNA levels in cultured adipocytes. FEBS (letters) 1996; 379: 55-59. [ Links ]
91. Ryan AS, Elahi D. The effects of acute hyperglycemia and hyperinsulinemia on plasma leptin levels: its relationship with body fat, visceral adiposity and age in women. J Clin Endocrinol Metab 1996; 81: 4433-4438. [ Links ]
92. Vidal H et al. The expression of ob gene is not acutely regulated by insulin and fasting in human abdominal subcutaneous adipose tissue. J Clin Invest 1996; 98: 251-255. [ Links ]
93. Widddowson et al. Acute hyperleptinemia does not modify insulin sensitivity in vivo in the rat. Hor Met Res 1998; 30: 259-262. [ Links ]
94. Mizuno et al. Leptin affects pancreatic endocrine gunctions through the sympathetic nervous system. Endocrinology 1998; 139: 3863-70. [ Links ]
95. Niki et al. Human obese gene: molecular screening in japanese and asian indian NIDDM patients associated with obesity. Diabetes 1996; 45: 675-678. [ Links ]
96. McGregor et al. Radioimmunological measurement of leptin in plasma of obese and diabetic human subjects. Endocrinology 1996; 137: 1501-1504. [ Links ]
97. Nolan et al. Effects of troglitazone on leptin production. Studies in vitro and in human subjects. Diabetes 1996; 45: 1276-1278. [ Links ]
98. Tuominen et al. Leptin synthesis is resistant to acute effects of insulin in IDDM patients. J Clin Endocrinol Metab 1997; 82: 381-382. [ Links ]
99. Verroti et al. Leptin levels in non-obese and obese children and young adults with type 1 DM. Eur J End 1998; 139: 49-53. [ Links ]
100. Bertin et al. Insulin and body fat distribution have no direct effect on plasma leptin levels in obese caucasian women with and without type 2 diabetes mellitus. Diab Met 1998; 24: 229-234. [ Links ]
101. Mizuno et al. Leptin affects pancreatic endocrine gunctions through the sympathetic nervous system. Endocrinology 1998;139: 3863-70. [ Links ]
102. Dandona P, Weinstock R, et al. Tumor necrosis factor-a in sera of obese patients: fall with weight loss. J Clin Endocrinol Metab 1998; 83: 2907-2909. [ Links ]
103. Halle M, Berg A Northoff H, Keul J. Importance of TNFa and leptin in obesity and insulin resistance: a hypothesis on the impact of physical exercise. Exerc Immunol Rev 1998; 4: 77-94. [ Links ]
104. Paolisso et al. Advancing age and insulin resistance: role of plasrna TNFa. Am J Physiol 1998; 275: E294-9. [ Links ]
105. Mantzoros et al. Cerebrospinal fluid in anorexia nervosa: correlation with nutritional status and potential role in resistance to weight gain. J Clin Endocrinol Metab 1997; 82: 1845-185 1. [ Links ]
106. Kopp et al. Serum leptin and body weight in females with anorexia and bulimia nervosa. Horm Met Res 1998; 30: 272-275. [ Links ]
107. Merabet et al. Increased plasma leptin concentrations in end stage renal disease. J Clin Endocrinol Metab 1997; 82:847-850 [ Links ]
108. Grundfeldt et al. Serum leptin levels in AIDS. J Clin Endocrinol Metab 1996; 81: 4342-4346. [ Links ]
109. Carr A et al. A syndrome of peripheral lipodistrophy, hyperlipidemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS 1998; 12: f5 1-58. [ Links ]
110. Mikhail et al. Leptin stimulates fetal and adult erytrhoid and myeloid development. Blood 1997; 89: 1507-1512. [ Links ]
111. Bennet et al. A role for leptin and its cognate receptor in hemotopoiesis. Cur Biol 1996; 6: 1170-80. [ Links ]